
Rare Case of SMA Linked With Myoclonic Epilepsy Detailed in Report
Written by |
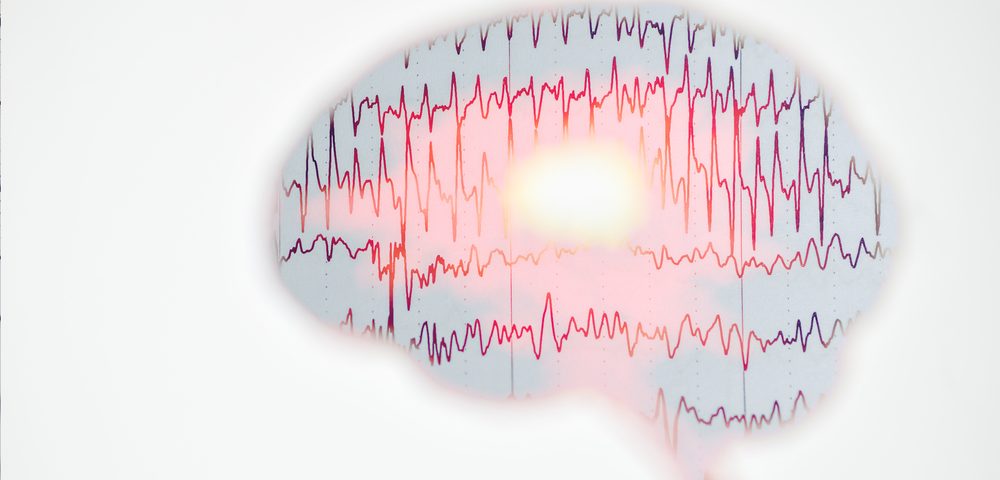
A 6-year-old boy was found to have a rare form spinal muscular atrophy (SMA) associated with progressive myoclonic epilepsy, a disease type known as “SMA plus” and not related to mutations in the causative gene for SMA, a case study reported.
SMA associated with progressive myoclonic epilepsy (SMA-PME) is a distinct disorder with its own clinical features, the researchers noted.
The case report, “Spinal Muscular Atrophy and Progressive Myoclonic Epilepsy: A Rare Association,” appeared in the Journal of Neurosciences in Rural Practice.
SMA is characterized by progressive weakness and atrophy of muscles due to loss of motor neurons. Most disease types are caused by mutations in the survival motor neuron 1 (SMN1) gene that encodes the SMN protein, which is essential for motor neuron survival.
In rare instances, however, SMA may be associated with progressive myoclonic epilepsy, a childhood seizure disorder that is unrelated to SMN genes. Progressive myoclonic epilepsy is characterized by progressive muscle weakness and atrophy that begins in childhood, followed by the development of myoclonic seizures — brief muscle “jerks” or spasms. SMA plus syndrome has previously been tied to mutations in the ASAH1 gene.
ASAH1 provides instructions to make an enzyme called acid ceramidase. This enzyme is usually found inside lysosomes — small, specialized cell compartments that digest and recycle different types of molecules — where it helps break down fatty molecules called ceramides.
Researchers in India described the case of a 6-year-old who began experiencing progressive muscle weakness and wasting in his limbs at age 2, delaying his motor milestones. Sudden, jerky muscle movements, characteristic of myoclonic seizures, were evident by the time he reached age 3.5. Months later, as a 4 year old, he had difficulty speaking and swallowing.
As the frequency of the muscle jerks increased, the boy developed postural tremors and twitches involving both hands, as well as periods of troubled awareness. A muscle examination found wasting in all limbs and diminished deep tendon reflexes, both characteristic of SMA.
Further analysis found abnormal electrical activity of the muscles, consistent with widespread loss of motor nerve connections. Electroencephalogram (EEG) showed brain activity consistent with generalized epilepsy.
Genetic analysis failed to identify SMN gene mutations, indicative of SMA with progressive myoclonic epilepsy not associated with the SMN gene.
The boy was treated with valproic acid and clobazam, which lessened myoclonic seizures and normalized EEG tests.
“To best of our knowledge, 44 cases of SMA-PME-like clinical presentation have been reported to date … Muscle weakness and wasting along with myoclonic jerks and seizures were common in all the reported cases of SMA-PME despite the differences in attributes like age of onset, severity and progression of the disease,” the researcher wrote.
“SMA with myoclonic epilepsy, unrelated to SMN gene, is a unique condition with its own clinical and electrophysiological features. Study of additional cases is also required to understand the genetic and pathophysiological [disease features] basis of SMA-PME,” they concluded.


Leave a comment
Fill in the required fields to post. Your email address will not be published.