

Spinal muscular atrophy overview
 Medically reviewed by Edward Smith, MD
Medically reviewed by Edward Smith, MD
Spinal muscular atrophy (SMA) is a rare genetic condition characterized by progressive muscle weakness and atrophy. It mainly affects motor function, but often also causes problems with speaking, swallowing, and breathing, along with other symptoms.
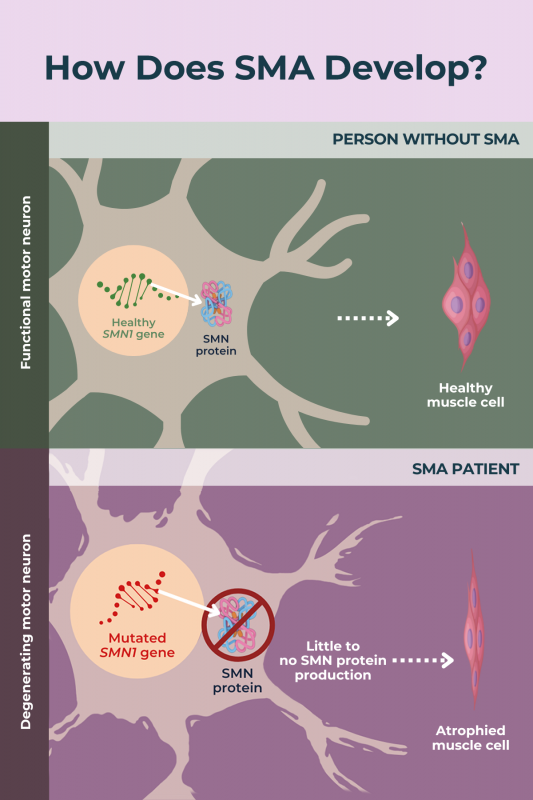
The most common types of SMA are caused by mutations in the SMN1 gene, leading to a deficiency of a protein called SMN, or survival motor neuron protein.
SMA is estimated to affect about 1 in every 10,000 live births, and about 1 in every 50 people is thought to be a carrier for the disease. Carriers are typically healthy individuals who have an SMA-causing mutation and can transmit it to their biological children.
SMA has been the most common genetic cause of death in infants and toddlers, although in the past decade the advent of new disease-modifying treatments has dramatically altered the prognosis for individuals with the disease.
What are the types of SMA?
SMA is divided into five main types based on the age at which symptoms first appear and whether or not children with the more severe forms of the disease hit motor development milestones.
Type 0
The rarest and most severe form of the disease, SMA type 0 is defined by symptoms that are apparent before birth, while the fetus is still in the womb. Babies with this type of SMA usually need medical assistance to keep them alive as soon as they are born, and most do not survive past the first 6 months of life.
Type 1
SMA type 1 is the most common form of SMA, and also one of the most severe, defined by symptoms that are apparent at birth or that develop within the first 6 months of life. In the absence of treatment, most children born with SMA type 1 will never be able to sit up unsupported, and most do not survive beyond age 2.
Type 2
The second most common type of SMA, type 2 disease is characterized by symptoms that appear between the ages of 6 and 18 months. Without treatment, most children with SMA type 2 are able to sit independently, but cannot stand or walk on their own and will often lose the ability to sit as they get older. Patients with this disease type often survive into adulthood, though life expectancy also is reduced without treatment.
Type 3
One of the milder forms of the disease, SMA type 3 is marked by symptoms that appear after 18 months of age during childhood or adolescence. It also is called juvenile SMA or Kugelberg-Welander syndrome. People with this form of SMA achieve the ability to walk independently, but without treatment many lose this ability as the disease progresses. Life expectancy seems to be unaffected in this type of SMA.
Type 4
SMA type 4, the mildest form of SMA and one of the rarest, is characterized by symptoms that appear in adulthood, usually after age 35. People with this form of SMA usually do not lose the ability to walk and life expectancy is unaffected.
Rarer forms
In addition to the five main types of SMA, all of which are due to mutations in the SMN1 gene located on chromosome 5, there are several other disease forms caused by different underlying genetic mutations. These SMA conditions also are characterized by muscle weakness and atrophy. Among them are:
- X-linked infantile SMA
- SMA with respiratory distress type 1 (SMARD1)
- SMA with lower extremity predominance (SMA-LED)
- SMA with progressive myoclonic epilepsy
- Finkel type SMA (SMAFK)
- spinal and bulbar muscular atrophy (SBMA), also called Kennedy’s disease
- distal spinal muscular atrophy (DSMA).
What are the causes of SMA?
The main types of SMA are caused by mutations in the SMN1 gene, which provides instructions for making the protein SMN. Due to mutations in SMN1, an inadequate amount of SMN protein is produced.
More like this...


The deficiency of SMN protein affects all the body’s cells, but motor neurons — the specialized nerve cells that control movement — are particularly sensitive. In SMA, the lack of SMN protein causes motor neurons to gradually sicken, or degenerate, and die off. As a result, muscles do not get the electrical signals that normally tell them to move, resulting in muscle weakness, ultimately leading to muscle atrophy over time.
How is SMA inherited?
Everyone inherits two copies of the SMN1 gene, one from each biological parent. SMA is an autosomal recessive disorder, meaning an individual will only develop the condition if both copies of SMN1 carry a disease-causing mutation.
People with only one mutated SMN1 copy are called carriers because they will not develop SMA themselves, but can pass the disease-causing mutation to their biological children. If two carriers conceive a child, there is a:
- 25% chance the child will have SMA
- 50% chance the child will be a carrier
- 25% chance the child will not have SMA and will not be a carrier.
What are the symptoms of SMA?
The hallmark symptom of SMA is muscle weakness, and it gets progressively worse over time. This weakness usually affects proximal muscles — muscles close to the torso, like those of the hips and shoulders — more profoundly than distal muscles in the hands and feet. Further, muscle weakness in SMA usually affects the lower limbs more than upper limbs.
Muscle weakness commonly causes challenges with mobility in people with the disease. Other common SMA symptoms and manifestations include:
- difficulty breathing
- fatigue
- scoliosis or an abnormal curvature of the spine
- hip dislocations and fracture-prone bones
- contractures or muscle tightening and hardening around a joint, limiting mobility
- gastroesophageal reflux disease, known as GERD, and other digestive issues
- tremors
- involuntary tongue movements
- difficulty with speech.
How is SMA treated?
Although there is no cure for SMA, approved therapies can slow or even stop progression of the disease. There also are many forms of supportive care that can help people with SMA stay healthier and live life to the fullest.
Medication
Disease-modifying therapies are treatments that have been proven in clinical trials to slow the progression of the disease. All of the approved SMA treatments work by boosting the production of the SMN protein.
While these therapies are effective in many cases for preventing SMA from worsening, their ability to reverse damage that has already happened is limited. As such, the best clinical outcomes are usually achieved when SMA treatment is started as early as possible.
In the U.S., four SMA treatments have received regulatory approval. The U.S. Food and Drug Administration (FDA) has approved these four therapies to treat all five main types of SMA (types 0, 1, 2, 3, and 4), though some are only indicated for use in certain age ranges.
- Evrysdi (risdiplam) is a daily oral therapy that can be taken by mouth or through a feeding tube. It is FDA-approved for patients of all ages.
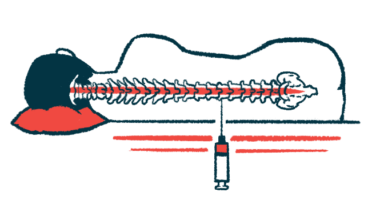
- Spinraza (nusinersen), administered via injection through the spine every four months, is FDA-approved for all ages.
- Zolgensma (onasemnogene abeparvovec-xioi), a one-time gene therapy, is FDA-approved for children up to age 2 years.
- Itvisma (onasemnogene abeparvovec-brve), also a one-time gene therapy, is FDA-approved for patients age 2 and older.
Nondrug treatments
In addition to medications that aim to slow disease progression, many people with SMA benefit from other forms of medical care and the use of specialized equipment. Some of these are:
- physical therapy (physiotherapy), which works to maintain and improve motor function
- occupational therapy, which aims to create strategies for navigating day-to-day life
- mobility equipment, ranging from wheelchairs to crutches to adaptive strollers, to help patients get around more easily and comfortably
- respiratory therapy and adaptive equipment to support breathing ability, which may include ventilators, assistive cough devices, and BiPAP (bi-level positive pressure) machines
- nutritional counseling and equipment such as feeding tubes to support adequate nutrition
- speech therapy to help with speaking difficulties as well as support chewing and swallowing abilities
- treatments to prevent and manage scoliosis, which can include postural therapy, the use of braces, or surgery in severe cases
- whole-body vibration therapy, which can help improve motor function and mobility
- emotional support, including counseling and support groups to manage mental health.
How is SMA diagnosed?
Because early treatment can slow disease progression and improve survival, a prompt SMA diagnosis is considered critical. All U.S. states and many other countries have implemented newborn screening programs, in which all infants are tested for SMA within the first few days of life. For older individuals, testing can be done at the time of symptom onset.
Genetic tests
The gold standard for diagnosing SMA is genetic testing, which is usually done on a small blood sample. This testing can identify the disease-causing mutations in the SMN1 gene or in other genes causing rarer SMA types.
Genetic testing also can identify the number of copies a patient has of the SMN2 gene — a backup gene that also provides instructions for making SMN protein. Most people have two copies of SMN2 but some have more. More copies of SMN2 are generally associated with less severe disease, and determining the number of SMN2 copies can be helpful in diagnosing SMA type.
Other tests
Before genetic testing became widely available, SMA was mainly diagnosed via assessments of muscle and motor neuron function. These include electromyography and nerve conduction tests — which measure the electrical activity in muscles and the motor neurons that control them — as well as muscle biopsies, where a small piece of muscle is taken for laboratory examination.
Today these tests are rarely included in diagnostic workups, but they can be useful in unusual cases in which the diagnosis is uncertain. Nowadays, genetic testing is the preferred method to confirm a diagnosis of SMA.
What is the prognosis of SMA?
The prognosis for SMA varies greatly depending on the disease type. Without treatment, patients with the severe types 0 and 1 don’t usually survive beyond infancy, while patients with type 2 disease often survive into adulthood. Life expectancy is usually unaffected in the milder SMA types 3 and 4.
Early treatment with disease-modifying therapies, all approved in the last decade, can now significantly alter the prognosis for babies with severe forms of the main types of SMA.
Babies with SMA who are treated early are increasingly reaching motor milestones, like sitting and walking, something that was not usually seen before medications were approved. Treatments also have been shown to slow disease progression and improve motor function in people with milder forms of SMA.
Because the first disease-modifying therapies for SMA only became available within the past 10 years — Spinraza was the first medicine for SMA to be approved in 2016 — long-term outcomes and life expectancy for people on these treatments are still unknown. Research is underway to better understand what the prognosis for SMA looks like in the modern era.
SMA News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.

FAQs about SMA
Category:
SMA Overview
There is no spinal muscular atrophy (SMA) cure. However, there are several approved therapies that can slow the progression of SMA and offer a better quality of life for people with the disease.
Category:
SMA Overview
Spinal muscular atrophy (SMA) is a progressive disease, meaning symptoms typically get worse as time goes on. There are multiple medications that have been proven to slow or even stop the progression of the main types of SMA. Apart from such medicines, other medical care strategies, including specialized equipment or physical therapy, can be of benefit to people with SMA and help make day-to-day life easier.
Category:
SMA Overview
Life expectancy for spinal muscular atrophy (SMA) varies by disease type — babies with the most severe forms of SMA usually don’t survive past infancy without treatment, while milder forms of the disorder do not usually affect life expectancy. Modern treatments have been shown to radically improve SMA life expectancy outcomes in severe disease types. However, because these therapies were only developed in the past decade, the long-term life expectancy for someone given modern treatment is still unknown.
Category:
SMA Overview
The gold standard for diagnosing spinal muscular atrophy (SMA) is genetic testing to identify disease-causing mutations. This testing can be done at any point in life, and it is even possible during pregnancy. It is recommended that babies be tested for SMA if there is a known family history of the disease, or if they show symptoms indicative of SMA, including muscle weakness, floppy limbs, difficulty feeding, trouble breathing, and failure to hit motor milestones like rolling over, sitting up, or crawling.
Category:
SMA Overview
According to the National Organization for Rare Disorders, spinal muscular atrophy (SMA) affects approximately 1 of every 10,000 live births, and the SMA Foundation estimates that about 10,000-25,000 children and adults in the U.S. have the disease. Because it affects fewer than 200,000 people in the country, SMA is considered a rare disease.
Related Articles
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion