Researchers Identify Process That Regulates How SMN2 Gene Is Read in SMA



Researchers have identified the process by which small compounds enable the production of the full SMN2 protein by effectively regulating how the gene is read, according to a study published in Nature Communications.
This finding suggests that these compounds may represent a new therapeutic option for spinal muscular atrophy (SMA), but also many other genetic diseases.
SMA is caused by lack of survival motor neuron (SMN) protein, which can be due to genetic mutations or deletion of its coding gene SMN1.
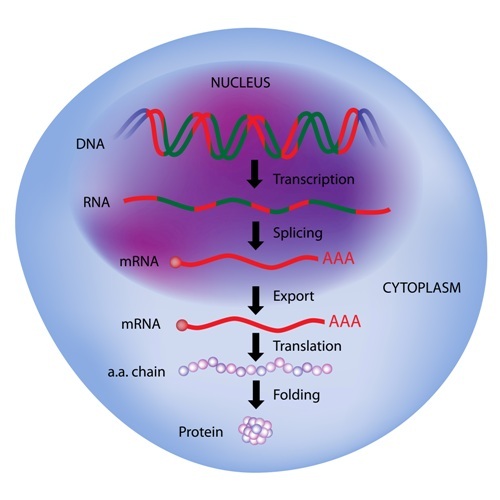
A second gene called SMN2 provides instructions for a protein that is very similar to SMN, but it misses one small part due to an exon-skipping process. Despite the fact that this smaller version of SMN is less stable and is unable to respond as well as the full-length version, is has been seen as a potential therapeutic target.
Identifying ways to prevent the skipping process to allow the correct reading of the full-length protein encoded by SMN2 can potentially overcome the lack of SMN that leads to the development of SMA.
Researchers have been trying to understand what exists in the SMN2 gene sequence that triggers the exon-skipping process, while at the same time looking at the cellular machinery involved in the process.
In a previous study several years ago, a team led by Friedrich Metzger, PhD, head of discovery rare diseases at Roche, showed that with very selective SMN2 skipping modifiers it was possible to inhibit the skipping process and promote the production of the full-length SMN protein encoded by the SMN2 gene.
In this more recent study, titled “Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers,” the team further explored the SMN2 skipping mechanism to better understand how the selective skipping (or splicing) modifiers that were previously identified actually work.
With the application of several techniques of genetic, biochemical, and chemical analysis, they found that the skipping modifiers would bind to two specific sites of the messenger RNA (mRNA) sequence (the product of the gene that will result in a protein) of SMN2. This would stabilize the mRNA molecule to allow the binding of other proteins responsible for the translation of the mRNA SMN2 sequence into a functional protein sequence.
“Our report suggests that a splicing deficit of an individual gene can be selectively targeted through specific small molecule interactions with RNA:protein complexes,” the researchers reported.
“As further findings highlight mis-splicing to be the cause of many disease-causing mutations, the work presented here has the potential to have widespread implications in the research and development of such RNA-targeting therapies,” they added.