
Glial Cells Key in Nerve Cell Communication That Helps Prevent Muscle Fatigue, Study Reports



A particular type of glial cell — non-neuronal cells of the nervous system — is important in communication between nerve cells and muscles, researchers report. The findings point to potential new targets for therapies that might prevent or lessen muscle fatigue in diseases like spinal muscular atrophy (SMA).
The study, “Activity-induced Ca2+ signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue,” was published in the journal eLife.
Unusual muscle fatigue is a feature of various clinical disorders, including those affecting motor nerve cells that control muscles. The progressive loss of these cells in SMA patients leads to muscular weakness and fatigue, and decreased endurance, limiting patients’ mobility.
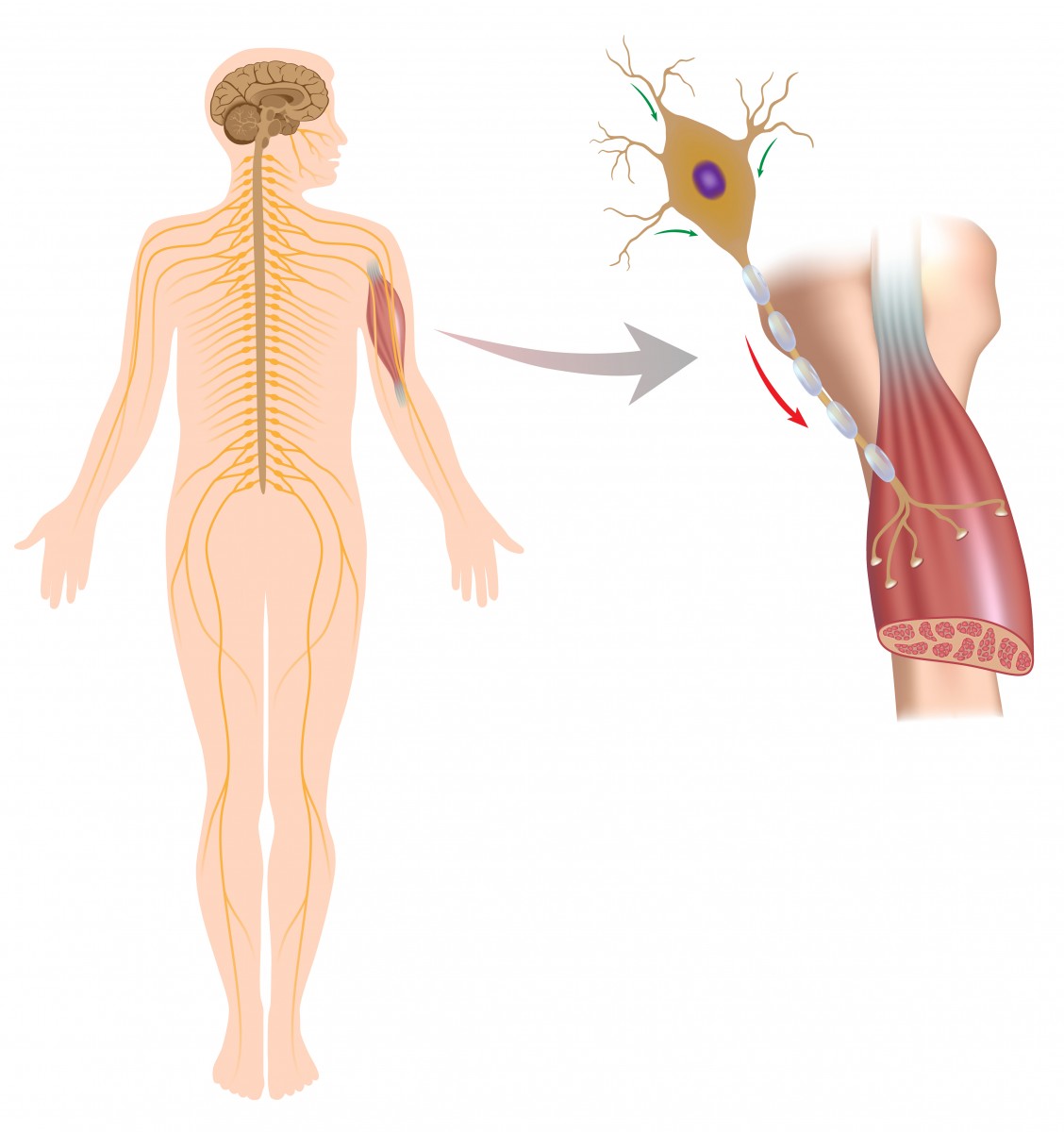
Muscle contraction results from the reaction of muscle cells to signals released by motor nerve cells. This takes place at a specialized site called the neuromuscular junction — the point of contact where a motor nerve cell reaches a muscle cell. Nerve cell failure to send the correct signals to muscles at these junctions is a cause of muscle fatigue.
An additional cell type present at the neuromuscular junction — called the terminal/perisynaptic Schwann cell (TPSC) — is a type of glial cell that covers the nerve-muscle contact area. These cells are known to have a role in the formation of neuromuscular junctions and in nerve cell repair, and respond to nerve cell activity by increasing their levels of calcium.
Communication problems between motor nerve and muscle cells can lead to muscle fatigue. Several studies have focused on the role of these particular cells in communication failures, but little is known about the role of TPSCs in these processes.
“Because these cells are activated by synaptic [nerve cell] activity, we wondered what the role of this activation was,” the study’s senior author, Thomas Gould with the University of Nevada, said in a news release by Ashley Yeager published in The Scientist.
To evaluate whether TPSCs were involved in muscle fatigue, researchers investigated these cells’ responses at the neuromuscular junction in mice genetically modified to mark calcium ions in these cells with a fluorescent light.
These responses were analyzed in the diaphragm muscle — the muscle that separates the thoracic cavity from the abdominal cavity — of young mice.
The stimulation of motor nerve cells in the diaphragm induced the release of calcium ions in TPSCs from their internal calcium stores, as well as the intake of potassium ions from the synaptic cleft, the narrow space between nerve and muscle cells where communication takes place.
However, this was not true for TPSCs lacking a specific protein, called the purinergic 2Y1 receptor (P2Y1R) protein, suggesting this protein somehow helps to mediate muscle fatigue.
Adding to this hypothesis was the fact that treatment with high levels of potassium induced greater muscle fatigue in mice lacking P2Y1R than in normal mice.
Calcium release “is believed to be a very important intracellular signal initiated by neural activity,” Gould said. The accumulation of potassium in the synaptic cleft — released by nerve and muscle cells during intense activity — also contributes to fatigue.
TPSCs seems to contribute to continued muscular contraction, preventing muscle fatigue by releasing calcium and taking up potassium. Therapeutic strategies to induce a greater calcium release by TPSCs — and thus increase their potassium uptake — could potentially help ease muscle fatigue in muscle-associated diseases, such as SMA.
The team plans to further determine the mechanisms underlying calcium release and potassium uptake in TPSCs, and to examine the role of other ions, in muscle fatigue.



