
2015 Researcher Meeting Promises Increased Understanding of SMA
Written by |
[Parameter-Settings] FileVersion = 2000 Date/Time = 0000:00:00 00:00:00 Date/Time + ms = 0000:00:00,00:00:00:000 User Name = TCS User Width = 1140 Length = 956 Bits per Sample = 8 Used Bits per Sample = 8 Samples per Pixel = 3 ScanMode = xy Series Name = 04.09.12.lei
Cure SMA, an organization dedicated to the treatment and cure of spinal muscular atrophy (SMA), recently revealed novel studies presented during its annual researcher meeting.
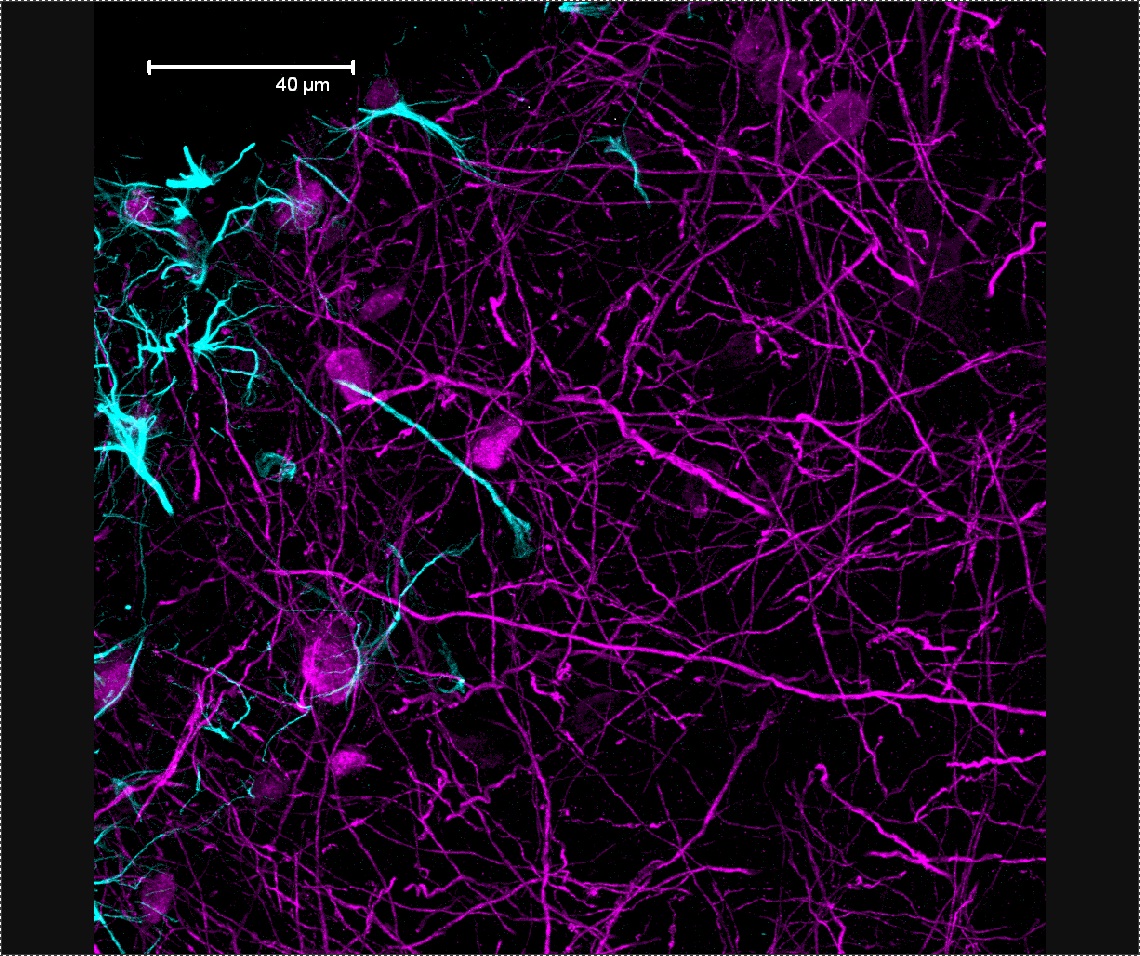
SMA is a genetically-determined neuromuscular disorder characterized by loss of specific spinal cord neurons, which causes weakness of the limbs and muscle degeneration. This leads to loss of voluntary muscle movement. The age of SMA onset, symptoms and rate of progression varies — the younger the onset, the more severe the symptoms. SMA is classified into types 1 through 4 to account for the variability in symptoms and onset age. Several types of SMA exist and each may be associated with different gene mutations. SMA is most commonly caused by a mutation of the SMN1 gene on chromosome 5. When the SMN1 gene is mutated, there is a deficit of a motor neuron protein called survival of motor neuron (SMN), a critical protein for motor neurons’ functionality.
Cure SMA supports research directed at restoring SMN expression, either through stimulating alternative genes to produce more SMA protein or by using gene therapy to correct the genetic mutation. Even alternative strategies that involve proteins other than SMA may be possible according to the foundation.
On their website, Cure SMA described four talks that occurred during their annual meeting:
- Sara Custer from the Androphy laboratory at Indiana University described newly discovered functions for SMN in neurons. According to Custer, SMN may control the development of brain cells (neurons) by interacting directly with alpha-COP, a protein located within cells. Alpha-COP may be another target that can be manipulated in treatments for SMA.
- Christine Beattie, from Ohio State University, further described how SMN may contribute to nervous system development, however based on its interaction with a different protein HuD. Beattie studies HuD in zebra fish, a commonly used experimental animal model.
- Seyyed Mohsen Hosseini-Barkooie from the Wirth lab at the University of Cologne, studies plastin-3 and SMN-depleted motor neurons. Increasing plastin-3 or coronin-1C increases motor neuron axonal development and helps motor neurons to function.
- Elena Bianchetti from the Pellizzoni lab at Columbia University studies the protein Stasimon in a mouse model of SMA. Stasimon is reduced when SMN protein levels are low. Bianchetti found that increasing SMN levels improves some SMA symptoms in mice.
The presentations illustrated several new potential protein targets for the treatment of SMA. Hopefully these early research efforts focusing on animal models will advance to human trials and eventual therapies for SMA.



Leave a comment
Fill in the required fields to post. Your email address will not be published.