
SMN2 Copy Number and Rare Variant Influence SMA Severity
Written by |

The number of copies of the “backup” SMN2 gene and a rare genetic variant in this gene influence disease severity in people with spinal muscular atrophy (SMA), according to data from more than 2,000 people involved in a free SMA genetic testing program.
Launched in 2018 by Biogen and Invitae Corporation, the program, called SMA Identified, resulted in a high diagnostic yield, further supporting the importance of genetic testing in individuals with a confirmed or suspected SMA diagnosis.
An early confirmed SMA diagnosis may speed access to clinical care, including disease-modifying therapies that may slow or prevent disease progression, the researchers noted.
Findings were published in the study, “SMA Identified: Clinical and Molecular Findings From a Sponsored Testing Program for Spinal Muscular Atrophy in More Than 2,000 Individuals,” in the journal Frontiers in Neurology.
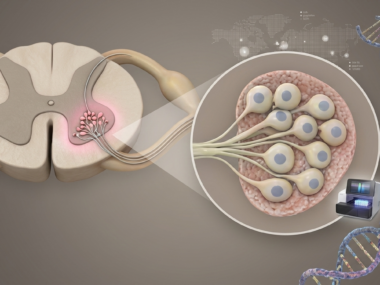
The most common genetic cause of death in infants and children, SMA is characterized by the progressive loss of motor neurons, the specialized nerve cells that control voluntary muscle movements, leading to muscle weakness and atrophy.
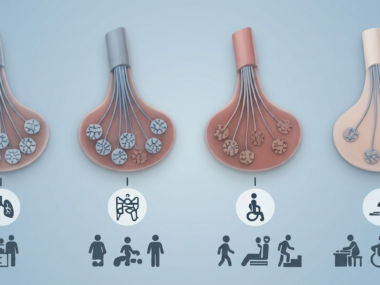
It is most often the result of mutations in both copies of the SMN1 gene, affecting the production of SMN, a protein essential for motor neuron and muscle health. The existence of a “backup” gene, SMN2, can partly compensate for the loss of SMN1-produced SMN. Typically, the more SMN2 gene copies a patient has, the less severe the disease,
People with only one defective SMN1 copy will not develop SMA, but will be carriers, meaning they can pass the mutation to their children.
Given the “narrow therapeutic window for optimally impacting progression of SMA … early molecular diagnoses are critical in managing patient care,” the researchers wrote, adding that such a diagnosis is also a requisite for approved treatments and clinical trials of SMA.
SMA Identified was designed to provide molecular diagnostic testing at no charge for individuals with clinical or suspected diagnoses of SMA, and to assess carrier status in first-degree relatives of people diagnosed with SMA.
The program is open to eligible individuals living in the U.S. and Puerto Rico, upon a request made by a qualified healthcare provider.
Biogen, which markets Spinraza (nusinersen), the first disease-modifying therapy approved for SMA, is providing financial support to the program, but does not receive any information on SMA patients identified through it.
In this study, researchers evaluated the diagnostic yield from SMA Identified and potential association between clinical features and positive genetic diagnoses.
Between March 2018 and March 2020, 2,459 unrelated individuals, with a mean age of 24.3 years, underwent genetic testing, which assessed SMN1 deletions and other mutations and quantified SMN2 copy number.
A positive result included the presence of either two disease-causing SMN1 mutations or a combination of a disease-causing SMN1 deletion (the classic cause of SMA) and a mutation of uncertain significance in SMN1 or SMN2.
A total of 771 individuals (31.3%), with a mean age of 25.7 years had a positive result, “demonstrating that a sponsored testing program is an effective approach for providing molecular diagnoses among individuals with a suspected or confirmed clinical diagnosis of SMA,” the researchers wrote.
Most (96.2%) positive diagnoses were attributed to the classic disease-associated deletion in both SMN1 copies, while 3.8% of cases carried one SMN1 deletion and one of 17 other SMN1 mutations.
Notably, individuals with mutations other than the classic deletion were significantly older at the time of testing, but had similar reported symptoms, than those with two classic deletions.
Still, these patients would “be missed by newborn screening programs,” which use genotyping technologies that can detect only classic SMN1 deletions, according to the researchers.
In addition, while muscle weakness was the most commonly reported symptom among participants, “it was not as predictive of a positive molecular diagnosis as was the presence of spinal rods, spinal fusions, and scoliosis,” the team added.
These findings “may provide clinicians additional guidance to systematically ascertain presenting symptoms that warrant genetic testing for SMA, which has important implications in the era of available therapies for SMA,” they wrote.
Moreover, among individuals with a positive SMA diagnosis, SMN2 copy number and the presence of a rare SMN2 mutation, called c.859G>C, were found to influence disease severity, measured by the number of reported symptoms and age at the time of testing.
The c.859G>C mutation is classified as a positive genetic modifier of SMA, as it increases SMN2-produced SMN and reduces disease severity.
In accordance, the 11 patients with a positive SMA diagnosis who also carried the SMN2 c.859G>C variant were older at the time of genetic testing, regardless of SMN2 copy number, further supporting an independent association between this variant and milder disease.
“Identifying SMA early through clinical features and confirming the diagnosis through genetic testing may have considerable impact by accelerating access to clinical care and reducing SMA-associated morbidity in affected individuals,” the researchers wrote.
They noted that the high diagnostic yield of SMA Identified may be related to greater awareness of the program and its free-of-charge nature, and that follow-up studies on the utility of such programs in providing earlier access to SMA treatment are required.
“Ongoing analysis of individuals tested through the SMA Identified program may provide additional insights into the clinical presentations most associated with a positive diagnosis, the genetic landscape of sequence variants in [patients with only one SMN1 deletion], and the influence of genetic modifiers on disease severity,” the team concluded.

Leave a comment
Fill in the required fields to post. Your email address will not be published.